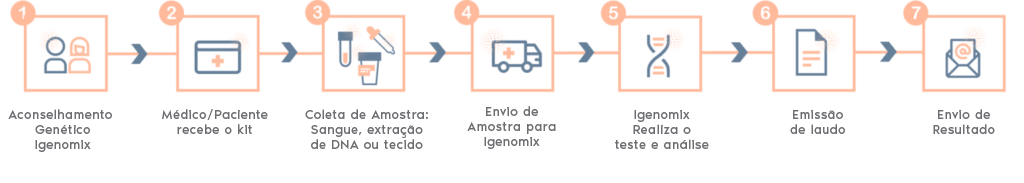
Painel de Precisão da Síndrome de Kallmann
A Síndrome de Kallmann (SK) é uma doença genética rara que pertence ao espectro do hipogonadismo hipogonadotrópico isolado. Essa diminuição da função gonadal se deve a uma falha na diferenciação ou migração de neurônios que surgem embriologicamente na mucosa olfatória e viajam para o hipotálamo.