Zenit by Nace
Aproximadamente uma em cada 80 gestações pode ser afetada por uma das condições genéticas examinadas pelo novo teste Zenit.
O Zenit representa uma redefinição do conceito atual de triagem pré-natal, pois analisa uma bateria abrangente de condições clínicas relevantes para o bem-estar do futuro bebê. Isso proporcionará maior tranquilidade para a paciente.
O Zenit oferece um novo valor agregado à avaliação de risco pré-natal, não contemplado pelas abordagens atuais.
- Além de avaliar o risco de aneuploidias cromossômicas no feto, o Zenit faz a triagem de 112
distúrbios de gene único de fenótipos moderados a graves com impacto significativo na qualidade de vida.
- Aumenta a taxa de detecção em um fator de cinco em comparação com outros testes básicos de triagem.
- Aumenta a taxa de detecção em um fator de três em comparação com o teste pré-natal não invasivo mais completo.

O Zenit by Nace oferece a abordagem clínica mais completa para:
- Triagem de condições citogenéticas: Telas para aneuploidias completas em todos os cromossomos e
deleções/duplicações com uma resolução comparável à do teste de cariótipo. - Triagem de mutações hereditárias: Avalia o risco de transmissão de mais de 100 condições recessivas.
Um painel de genes recomendado pelo American College of Medical Genetics and Genomics (ACMG).
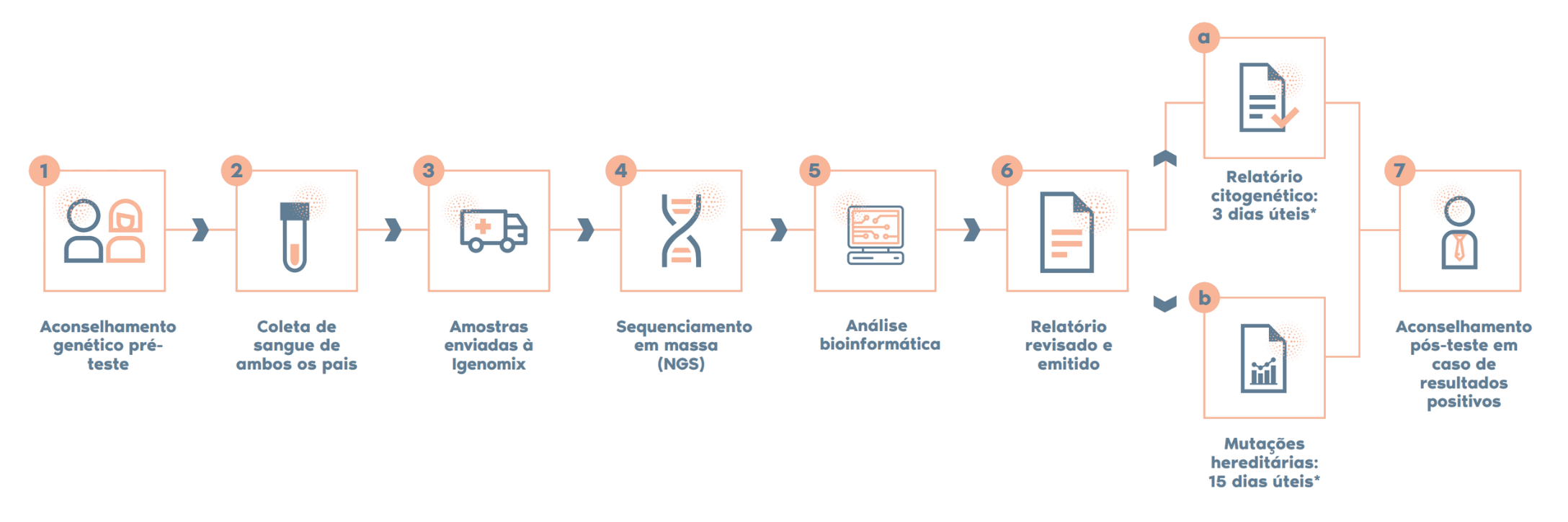
Como é o procedimento?